Quand un médicament générique doit être approuvé, les autorités sanitaires ne se contentent pas de vérifier que les ingrédients sont les mêmes. Elles exigent une preuve concrète : le produit générique doit se comporter dans le corps comme le médicament d’origine. C’est ce qu’on appelle l’équivalence bioéquivalente (BE). Mais pour certains médicaments, cette évaluation devient un défi technique majeur. Ce sont les drugs hautement variables (HVD) - des substances comme la warfarine, la levothyroxine ou le prasugrel - dont la réponse dans l’organisme varie énormément d’un patient à l’autre. Pour ces cas, la méthode classique de comparaison (deux périodes, deux traitements) échoue. C’est là que les études réplicatives entrent en jeu.
Pourquoi les études classiques ne suffisent plus
Dans une étude bioéquivalente standard, chaque participant reçoit d’abord le médicament générique, puis après une période de lavage, le médicament de référence. On mesure la concentration dans le sang à différents moments. On calcule ensuite deux paramètres clés : l’aire sous la courbe (AUC) et la concentration maximale (Cmax). Si ces valeurs sont proches entre les deux produits - typiquement entre 80% et 125% -, on conclut à l’équivalence. Mais cette règle tombe à l’eau avec les HVD. Pourquoi ? Parce que la variabilité intra-sujet (ISCV) dépasse 30%. Cela signifie qu’un même patient, sous le même traitement, peut montrer des concentrations plasmatiques très différentes d’une prise à l’autre. Avec une ISCV de 40% ou 50%, la marge de 80-125% devient trop large pour garantir la sécurité. On risque d’approuver un générique qui n’est pas vraiment équivalent. Ou pire : rejeter un bon produit parce que la variabilité naturelle du médicament fausse les résultats. Les études classiques exigent alors des centaines de sujets pour avoir une puissance statistique suffisante. Des études avec 98 participants qui échouent malgré un investissement colossal - c’est une réalité courante avant l’arrivée des designs réplicatifs.Comment fonctionnent les études réplicatives ?
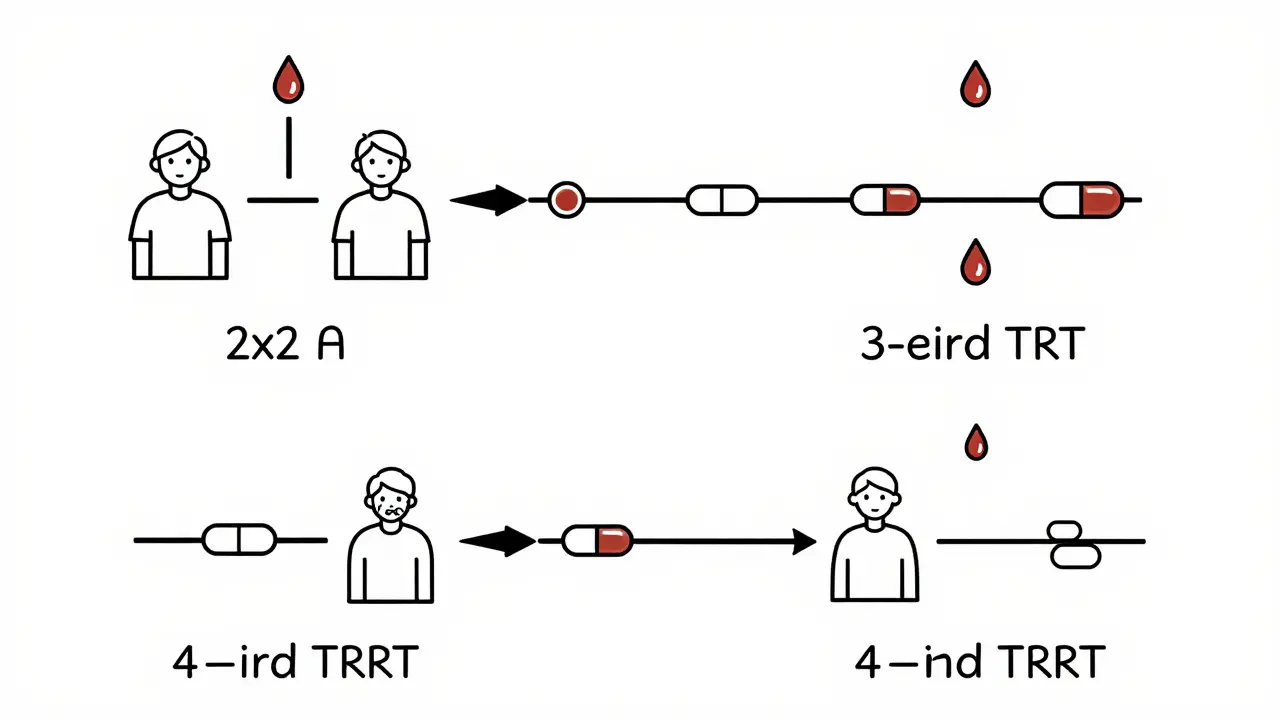
Les études réplicatives changent la donne en permettant de mesurer la variabilité de chaque formulation. Au lieu de ne recevoir qu’un seul traitement par période, les participants reçoivent plusieurs fois les deux produits, dans des séquences différentes. Il existe trois types principaux :- Design complet réplicatif (4 périodes) : TRRT ou RTRT. Chaque sujet reçoit le test (T) deux fois et le référence (R) deux fois. Cela permet d’estimer la variabilité intra-sujet pour les deux produits (CVwT et CVwR).
- Design partiellement réplicatif (3 périodes) : TRR, RTR, RRT. Ici, le produit de référence est administré deux fois à chaque sujet, mais le générique seulement une fois. On ne peut estimer que la variabilité du produit de référence (CVwR).
- Design complet réplicatif (3 périodes) : TRT ou RTR. Chaque sujet reçoit T une fois et R deux fois (ou inversement). C’est le plus utilisé en pratique aujourd’hui.
La clé ? Ces designs permettent d’appliquer la réduction de l’échelle de référence (RSABE). Au lieu de comparer les moyennes avec une limite fixe (80-125%), on étend cette limite en fonction de la variabilité observée du produit de référence. Plus la variabilité est élevée, plus la marge s’élargit - mais toujours dans des limites de sécurité strictes définies par les agences.
Par exemple, si le CVwR est de 45%, la limite d’équivalence peut passer à 69,8%-143,2%. Cela rend l’évaluation possible sans avoir besoin de 120 sujets. Avec un design réplicatif, on obtient la même puissance statistique avec seulement 24 à 48 participants.
Quel design choisir ?
Le choix du design n’est pas anodin. Il dépend de la variabilité attendue du médicament.- ISCV < 30% : Restez sur une étude classique 2x2. C’est plus simple, moins coûteux, et suffisant.
- ISCV entre 30% et 50% : Optez pour un design complet réplicatif en 3 périodes (TRT/RTR). C’est l’équilibre idéal entre puissance statistique et faisabilité opérationnelle. 83% des CRO interrogés en 2023 l’ont choisi comme standard.
- ISCV > 50% : Un design en 4 périodes (TRRT/RTRT) est recommandé, surtout pour les médicaments à indice thérapeutique étroit (NTI) comme la warfarine. La FDA exige ce design pour ces cas précis.
La FDA et l’EMA ont des exigences légèrement différentes. L’EMA accepte les designs partiellement réplicatifs (TRR/RTR/RRT) pour les HVD, mais exige que 12 sujets au moins fournissent des données depuis la séquence RTR. La FDA, elle, préfère les designs complets pour les NTI, mais accepte les designs partiels pour les autres HVD.
Les avantages concrets : moins de sujets, plus de succès
Les chiffres parlent d’eux-mêmes. Dans une étude de 2020, une ISCV de 50% et une différence de formulation de 10% nécessitaient 108 sujets pour une étude classique. Avec un design réplicatif, il en fallait seulement 28. Une réduction de 74%.En 2023, 68% des études BE pour HVD aux États-Unis utilisaient un design réplicatif, contre 42% en 2018. Le taux d’approbation pour ces études est de 79%, contre 52% pour les études classiques. Dans les données de l’EMA, 78% des génériques HVD approuvés en 2023 ont été évalués via un design réplicatif - et 63% d’entre eux utilisaient le modèle TRT/RTR.
Un manager clinique sur le forum BEBAC a rapporté que son étude sur la levothyroxine, menée avec 42 sujets en design TRT/RTR, a été approuvée du premier coup. Une précédente tentative avec 98 sujets en 2x2 avait échoué.
Les pièges à éviter
Ce n’est pas une solution magique. Les études réplicatives introduisent de nouvelles complexités.- Retraits des participants : Dans les études à 4 périodes, les taux de retrait peuvent atteindre 25%. Il faut prévoir 20-30% de sur-recrutement. Un statisticien sur Reddit a dû étendre son étude de 8 semaines et dépenser 187 000 $ de plus après un taux de retrait de 30%.
- Périodes de lavage insuffisantes : Pour les médicaments à demi-vie longue, un lavage de 7 jours ne suffit pas. Une contamination résiduelle fausse les mesures.
- Modèles statistiques mal choisis : Il ne suffit pas de copier un script R. Il faut comprendre les modèles à effets mixtes, la réduction d’échelle, et les hypothèses sous-jacentes. L’outil replicateBE (version 0.12.1) est devenu le standard, avec plus de 1 200 téléchargements au premier trimestre 2024.
La formation est essentielle. Selon un atelier AAPS en 2022, il faut entre 80 et 120 heures de formation spécialisée pour maîtriser l’analyse correcte.

Le futur : harmonisation et intelligence artificielle
L’harmonisation mondiale est en cours. L’ICH travaille sur une mise à jour de l’E14/S6(R1) prévue pour le troisième trimestre 2024. La FDA propose de standardiser les designs 4-périodes pour tous les HVD avec ISCV > 35%. L’EMA reste plus flexible. Cette divergence crée encore des rejets : 23% des soumissions à l’EMA utilisant un design FDA préféré ont été rejetées en 2023.Des innovations émergent. Pfizer a testé en 2023 un modèle d’apprentissage automatique qui prédit avec 89% de précision le nombre de sujets nécessaires, en se basant sur des données historiques. La FDA a aussi accepté en mai 2023 l’usage de méthodes bayésiennes pour certaines études réplicatives - une première.
Le marché mondial des études BE a atteint 2,8 milliards de dollars en 2023. Les études réplicatives représentent 35% des évaluations HVD, contre 18% en 2019. La croissance annuelle est de 15,3% jusqu’en 2028. Les grands CRO comme WuXi AppTec, PPD et Charles River dominent, mais les spécialistes comme BioPharma Services gagnent du terrain grâce à leur expertise statistique.
Comment commencer ?
Si vous travaillez sur un HVD :- Estimez la variabilité attendue du produit de référence (CVwR) à partir des données de littérature ou d’études antérieures.
- Si CVwR < 30% : utilisez un design 2x2.
- Si CVwR entre 30% et 50% : choisissez un design TRT/RTR en 3 périodes.
- Si CVwR > 50% ou si c’est un NTI : optez pour TRRT/RTRT en 4 périodes.
- Recrutez 20-30% de sujets supplémentaires pour compenser les départs.
- Formez votre équipe statistique à l’analyse RSABE avec replicateBE ou Phoenix WinNonlin.
- Consultez les guidances spécifiques de la FDA et de l’EMA pour votre médicament.
Les études réplicatives ne sont pas une mode. Ce sont devenus la norme pour les HVD. Elles permettent de faire des études réalisables, rigoureuses, et éthiques - sans sacrifier la sécurité des patients. Ignorer cette méthode, c’est risquer un échec coûteux, un retard de mise sur le marché, ou pire : un générique qui ne fonctionne pas comme il le devrait.
Quelle est la différence entre un design réplicatif et un design classique en bioéquivalence ?
Dans un design classique (2x2), chaque participant reçoit le produit test et le produit de référence une seule fois chacun, dans un ordre croisé. Dans un design réplicatif, les participants reçoivent plusieurs fois les deux produits - par exemple, test, référence, référence (TRR) ou test, référence, test (TRT). Cela permet de mesurer la variabilité intra-sujet de chaque formulation, ce qui est impossible avec un design classique. Cette mesure est essentielle pour les médicaments hautement variables (HVD), car elle permet d’ajuster les limites d’équivalence selon la variabilité réelle, rendant l’étude plus fiable et moins coûteuse.
Pourquoi les études réplicatives sont-elles obligatoires pour les médicaments à indice thérapeutique étroit (NTI) ?
Les NTI, comme la warfarine ou la levothyroxine, ont une marge de sécurité très étroite. Une petite variation dans la concentration plasmatique peut entraîner un échec thérapeutique ou un effet toxique. Les études réplicatives permettent d’estimer avec précision la variabilité du produit de référence et du générique. Cela permet d’appliquer une échelle de réduction (RSABE) qui garantit que le générique n’est pas seulement « proche » du produit d’origine, mais qu’il est aussi stable et prévisible dans son comportement. Sans cette précision, l’approbation d’un NTI serait trop risquée.
Quel est le design réplicatif le plus utilisé aujourd’hui ?
Le design complet réplicatif en 3 périodes (TRT/RTR) est devenu le standard dans l’industrie. Il offre un bon équilibre entre puissance statistique et faisabilité opérationnelle. Il permet d’estimer la variabilité du produit de référence (CVwR) avec une précision suffisante pour appliquer la RSABE, tout en nécessitant moins de périodes et de sujets qu’un design en 4 périodes. En 2023, 83% des CRO interrogés l’ont jugé optimal pour les HVD avec une ISCV entre 30% et 50%.
Quels logiciels sont utilisés pour analyser les données des études réplicatives ?
Les deux outils principaux sont Phoenix WinNonlin (commercial) et le package R replicateBE (gratuit et open-source). Le package replicateBE est devenu le standard de facto dans les laboratoires académiques et les CRO, avec plus de 1 200 téléchargements au premier trimestre 2024. Il permet d’implémenter les modèles RSABE selon les guidances FDA et EMA, y compris les modèles à effets mixtes, les estimations de CVwR, et les tests de réduction d’échelle. Les analystes doivent être formés à ces outils, car les méthodes statistiques sont beaucoup plus complexes que pour les études classiques.
Quels sont les principaux défis opérationnels des études réplicatives ?
Les trois principaux défis sont : 1) Le taux de retrait des participants, qui peut atteindre 25% dans les études à 4 périodes, obligeant à sur-recruter de 20 à 30%. 2) La longueur des périodes de lavage, particulièrement problématique pour les médicaments à demi-vie longue. 3) La complexité de l’analyse statistique, qui exige des compétences spécialisées en modèles à effets mixtes et en réduction d’échelle. Une mauvaise modélisation peut fausser les résultats et entraîner un rejet réglementaire. La formation des équipes est donc cruciale.