En 2026, les médicaments génériques représentent 65 % des prescriptions en Europe, mais seulement 18 % de la valeur totale du marché. Pourtant, leur accès n’est pas uniforme. Un même médicament peut être disponible en Allemagne six mois avant qu’il ne le soit en Bulgarie, malgré des règles communes. Ce décalage n’est pas un accident : il est le résultat d’un système réglementaire complexe, à plusieurs voies, qui cherche à équilibrer concurrence, sécurité et efficacité. Et en 2025, tout a changé.
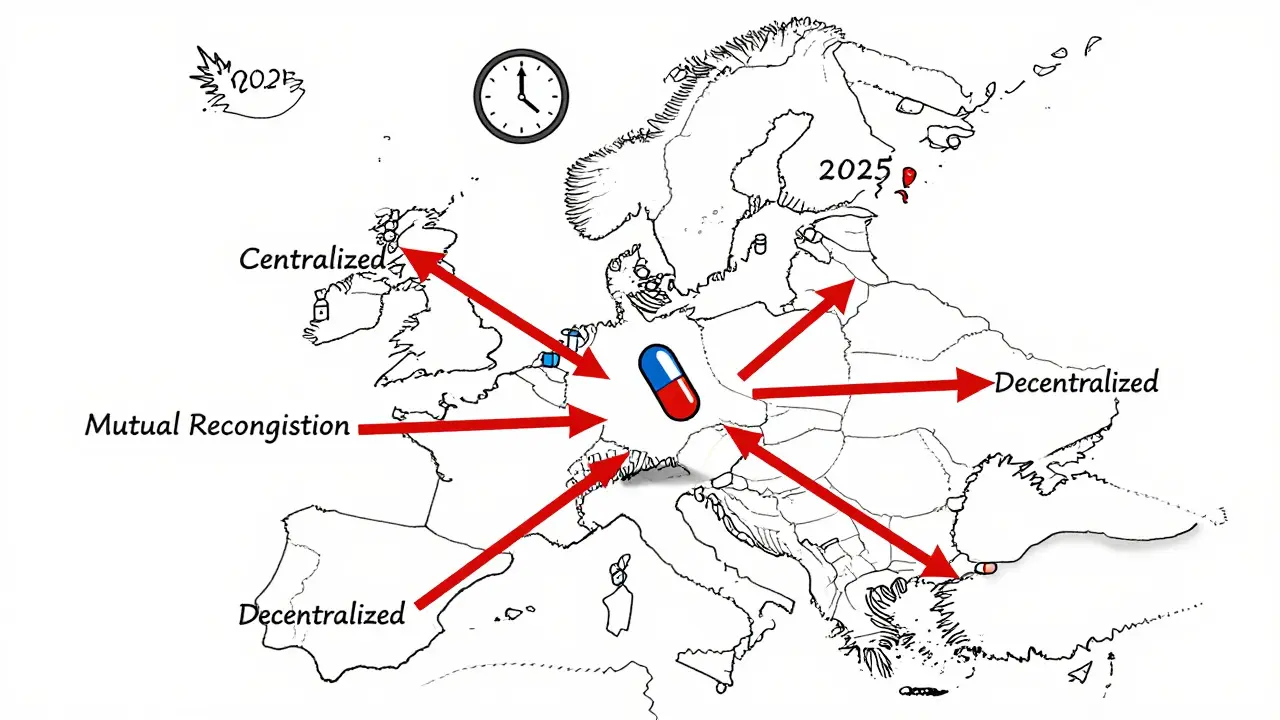
Quatre voies, un même objectif
En Europe, un fabricant de génériques ne peut pas simplement déposer une demande unique et attendre que l’Europe entière l’accepte. Il doit choisir parmi quatre voies d’autorisation, chacune avec ses avantages, ses coûts et ses pièges.La procédure centralisée (CP) est la plus rapide, mais aussi la plus chère. Elle permet d’obtenir une autorisation valable dans les 27 pays de l’UE, plus l’Islande, le Liechtenstein et la Norvège. L’Agence européenne des médicaments (EMA) évalue la demande en 180 jours (au lieu de 210 avant 2025). Le coût ? Entre 425 000 € et 2 millions d’euros. Ce n’est rentable que pour les génériques de très haut volume, comme ceux qui visent des médicaments de plus de 250 millions d’euros de ventes annuelles en Europe. Sandoz l’a utilisé pour son générique de Cosentyx : il a été lancé simultanément dans toute l’UE, 11 mois plus vite que par les voies traditionnelles.
La procédure de reconnaissance mutuelle (MRP) est la plus utilisée, avec 42 % des demandes. Elle commence par une autorisation dans un pays (l’État de référence), puis les autres pays doivent l’accepter. En théorie, ça prend 90 jours. En pratique, ça dure 132 jours en moyenne. Pourquoi ? Parce que chaque pays peut demander des tests supplémentaires. Teva a perdu 8,2 mois sur son générique de rosuvastatin à cause de négociations de prix en Allemagne qui ont bloqué l’accès aux Pays-Bas et en Belgique.
La procédure décentralisée (DCP) permet de déposer simultanément dans plusieurs pays sans autorisation préalable. Elle concerne 38 % des demandes. Mais elle est devenue le point noir du système. 37 % des dossiers connaissent des retards de plus de six mois, surtout dans les pays d’Europe de l’Est, où les exigences en matière de qualité varient d’un pays à l’autre. Les fabricants perdent des mois à répondre à des demandes contradictoires.
Enfin, la procédure nationale (5 % des cas) est réservée aux cas spécifiques : un générique destiné uniquement à un seul marché, souvent parce qu’il est trop niche pour justifier une approche européenne. Mais même là, les délais sont longs : entre 180 et 240 jours. Accord Healthcare a mis 197 jours pour obtenir une autorisation en France, contre 142 jours en MRP pour cinq pays à la fois.
La réforme de 2025 : un coup de tonnerre
Le 4 juin 2025, l’UE a adopté le « Pharma Package », la plus grande réforme réglementaire des génériques en 20 ans. Elle ne change pas seulement les règles - elle change la logique même du marché.La première révolution ? L’exemption Bolar étendue. Avant 2025, les fabricants pouvaient entamer les négociations de prix et de remboursement seulement deux mois avant l’expiration du brevet. Désormais, ils peuvent le faire six mois avant. Cela réduit de 4,3 mois en moyenne le délai entre l’expiration du brevet et le lancement du générique. Cela signifie aussi que les hôpitaux et les assureurs peuvent négocier plus tôt - ce qui pousse les prix à la baisse dès le départ. Selon REMAP Consulting, cela pourrait réduire les prix de 12 à 18 % au lancement.
Deuxième changement majeur : la protection des données. Avant, les données cliniques d’un médicament original étaient protégées 10 ans. Désormais, c’est 8 ans, avec 1 an de protection de marché. Et si le médicament répond à un besoin de santé publique, cette période peut être prolongée à 2 ans. Pour les fabricants de génériques, c’est une avancée : ils peuvent entrer plus tôt sur le marché. Mais pour les entreprises moyennes, cela pose un problème : les « bons de exclusivité transférables » (valables à partir de 490 millions d’euros de ventes) favorisent les géants. Les petits acteurs n’ont pas les moyens de jouer dans cette cour.
Troisième changement : l’obligation d’approvisionnement. Désormais, les fabricants doivent garantir un approvisionnement suffisant. Mais ici, le système est encore brouillé. Chaque pays définit « suffisant » à sa manière. Certains exigent des stocks de six mois, d’autres seulement trois. Ce manque de clarté pourrait créer des pénuries artificielles, surtout dans les petits marchés.

Les défis techniques : bioéquivalence et complexité
Pour qu’un générique soit approuvé, il doit être identique à l’original en composition, forme et effet. Mais « identique » ne veut pas dire « simple ». Pour les comprimés, c’est facile. Pour les inhalateurs, les crèmes, les solutions injectables complexes, c’est un défi.Les exigences de bioéquivalence sont strictes : les valeurs de Cmax (concentration maximale) et d’AUC (exposition totale) doivent se situer entre 80 % et 125 % de celles du médicament de référence. Mais certains pays vont plus loin. L’Allemagne exige des études pharmacodynamiques supplémentaires pour les inhalateurs. La France demande des données spécifiques sur les formulations pédiatriques. Le résultat ? 68 % des fabricants interrogés en 2025 citent des exigences nationales incohérentes comme leur principal obstacle.
Et puis il y a les polymorphes. Certains principes actifs peuvent exister sous plusieurs formes cristallines. La forme la plus stable est celle qui est brevetée. Les génériques doivent prouver qu’ils utilisent la même forme. L’Allemagne exige des données de stabilité supplémentaires. La France, elle, demande des tests de dissolution spécifiques. Sans ces détails, le dossier est rejeté.
Qui domine le marché ?
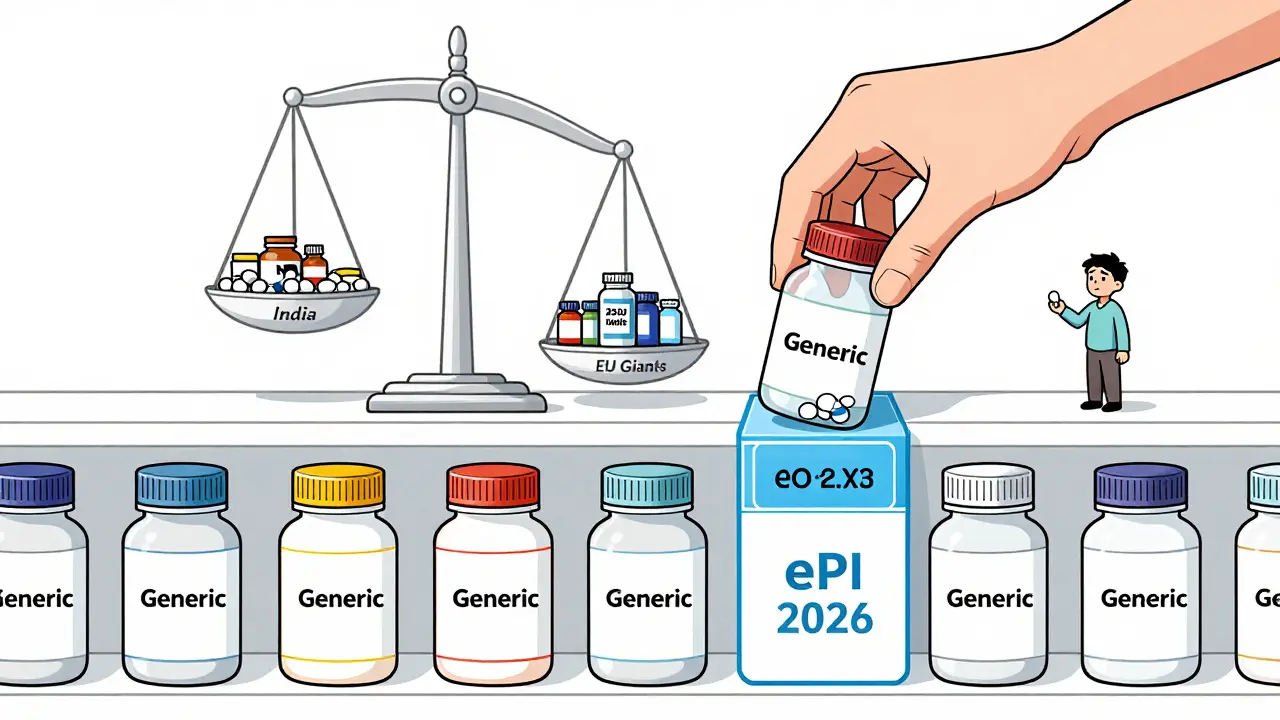
En 2024, les fabricants indiens ont obtenu 38 % des autorisations de génériques en Europe, contre 29 % en 2020. Ils gagnent du terrain grâce à des coûts de production bas et une grande agilité dans les procédures nationales.En revanche, les entreprises européennes comme Sandoz et Viatris conservent 52 % du marché. Comment ? En utilisant la procédure centralisée pour les produits à fort volume. Elles investissent dans les outils numériques, les équipes spécialisées, et elles anticipent les changements réglementaires.
Le marché européen des génériques vaut 42,7 milliards d’euros en 2024. Il pousse à 69,2 % de parts de marché d’ici 2028. Mais cette croissance ne sera pas équitable. Les pays du Sud et de l’Est voient des taux de croissance de 9,8 % par an. Les marchés plus matures, comme la France ou l’Allemagne, progressent à moins de 4 %. La réforme de 2025 pourrait creuser cette fracture.
Les nouvelles exigences numériques
À partir de 2026, tous les documents d’autorisation doivent être soumis en format XML, via le système ePI (informations sur le produit électronique). Cela signifie que chaque fabricant doit moderniser ses systèmes informatiques. Le coût ? Entre 180 000 et 250 000 € par entreprise. Pour les petites sociétés, c’est une barrière. Pour les grandes, c’est une opportunité : automatiser les soumissions réduit les erreurs, accélère les retours, et permet de réagir plus vite aux demandes des autorités.Le portail de l’EMA « Questions et réponses sur les génériques » est mis à jour chaque trimestre. Mais 58 % des fabricants disent que les réponses des autorités nationales contredisent souvent les lignes directrices de l’EMA, surtout sur les impuretés dans les médicaments anciens. C’est un chaos. Une entreprise peut obtenir une autorisation en Espagne, puis se voir refuser la même chose en Pologne pour une impureté identique.
Que reste-t-il à faire ?
La réforme de 2025 a ouvert la voie à un marché plus dynamique. Mais elle n’a pas résolu les problèmes structurels : la fragmentation, les incohérences nationales, les coûts élevés de conformité. Le vrai défi, d’ici 2028, sera de transformer cette architecture en un système réellement harmonisé - pas seulement en théorie, mais en pratique.Les génériques sont la clé pour réduire les coûts de santé. Mais pour qu’ils soient accessibles partout, l’Europe doit arrêter de laisser chaque pays faire à sa manière. Les patients n’ont pas besoin de 27 systèmes différents. Ils ont besoin d’un seul système fiable, rapide et juste.
Quelle est la différence entre la procédure centralisée et la procédure de reconnaissance mutuelle ?
La procédure centralisée donne une autorisation valide dans toute l’UE en une seule demande, évaluée par l’EMA. C’est rapide, mais très coûteux. La reconnaissance mutuelle commence par une autorisation dans un seul pays, puis les autres pays doivent l’accepter. C’est moins cher, mais plus lent et plus aléatoire, car chaque pays peut imposer des exigences supplémentaires.
Pourquoi les génériques sont-ils plus lents à arriver en Europe qu’aux États-Unis ?
En moyenne, il faut 22,4 mois pour qu’un générique soit disponible en Europe après son lancement aux États-Unis, contre seulement 8,7 mois entre les États-Unis et le Canada. Cette différence vient de la complexité du système européen : plusieurs voies d’autorisation, des exigences nationales variées, et des négociations de prix qui ralentissent tout.
La réforme de 2025 va-t-elle faire baisser les prix des génériques ?
Oui, mais pas immédiatement. L’extension de l’exemption Bolar permet aux fabricants de négocier les prix plus tôt, ce qui augmente la pression concurrentielle dès le lancement. Selon les modèles économiques, cela pourrait réduire les prix de 12 à 18 % au moment du lancement. À long terme, cela devrait aussi augmenter la concurrence, ce qui maintiendra les prix bas.
Quels pays sont les plus difficiles pour lancer un générique ?
L’Allemagne, la France et l’Italie sont les plus exigeants en termes de documentation. L’Allemagne demande des études supplémentaires pour les inhalateurs, la France exige des données pédiatriques, et l’Italie a des exigences strictes sur les impuretés. Les pays d’Europe de l’Est, comme la Pologne ou la Roumanie, posent des problèmes de cohérence : les mêmes dossiers peuvent être acceptés dans un pays et rejetés dans un autre.
Comment les fabricants indiens réussissent-ils à conquérir le marché européen ?
Ils se concentrent sur les procédures nationales et décentralisées, où les coûts sont plus bas et les délais plus courts. Ils investissent dans des équipes locales, comprennent les spécificités nationales, et utilisent des partenaires européens pour les aider à naviguer dans les systèmes. Leur agilité leur permet de répondre plus vite que les grands groupes occidentaux.




15 commentaires
Guy COURTIEU
C’est fou comment on peut avoir 65 % des prescriptions mais seulement 18 % de la valeur… On dirait que les gens veulent des génériques, mais qu’on les pousse à payer plus cher pour des marques. 🤦♂️
Floriane Jacqueneau
La réforme de 2025 est un pas dans la bonne direction, mais il faut reconnaître que la fragmentation nationale reste un cauchemar. L’Allemagne exige des études pharmacodynamiques pour les inhalateurs, la France veut des données pédiatriques, et l’Italie bloque sur les impuretés… On est dans un système qui punit l’efficacité.
Quentin Tridon
Sandoz a utilisé la procédure centralisée pour Cosentyx ? Bien sûr. Moi je dis : si tu veux jouer dans la cour des géants, faut avoir un budget de 2 millions d’euros. Les PME ? Elles se contentent des miettes. #CapitalismeEnModeGénérique 😎
Cyrille Le Bozec
Toute cette paperasse c’est juste une façon de protéger les big pharma et les labos allemands. Les Indiens ils arrivent avec des prix à 30 % et on les bloque avec des tests de dissolution et des polymorphes. C’est pas de la science c’est du protectionnisme déguisé
Léon Kindermans
Et si je te disais que l’EMA et les autorités nationales sont en train de créer des pénuries artificielles pour faire monter les prix ? Les stocks de 3 mois vs 6 mois ? C’est pas un hasard. C’est orchestré. Qui profite de ça ? Les multinationales. Et toi tu penses que c’est pour la sécurité ? 🤔
Marvin Goupy
Le vrai problème ? L’absence de standardisation des données. XML c’est bien, mais si l’Espagne accepte un impureté que la Pologne refuse, on est dans le chaos. 58 % des fabricants disent que les réponses nationales contredisent l’EMA. C’est pas un bug, c’est un feature.
Jean-Marc Frati
J’adore quand les gens disent que les génériques c’est moins cher… Mais oublie pas que derrière chaque générique il y a un ingénieur qui a passé 2 ans à prouver que sa forme cristalline est identique à la tienne. Le vrai héros ? Le chimiste de Bangalore qui bosse 18h par jour pour que tu puisses acheter ton rosuvastatin à 3€. 🙌
mathilde rollin
Je trouve ça vraiment important de rappeler que les patients ne demandent pas un système européen complexe. Ils veulent juste un médicament sûr, disponible, et abordable. Peu importe qu’il vienne d’Inde ou de Suisse. L’essentiel, c’est la santé.
nadine deck
La réforme de 2025, bien que bien intentionnée, risque d’accentuer les inégalités entre les grandes entreprises et les acteurs indépendants. La protection des données réduite à 8 ans, couplée aux bons de transfert à 490 millions d’euros, crée un monopole déguisé. Il faut une régulation plus équitable.
cyril le boulaire
Je suis allé voir un médecin hier. Il m’a prescrit un générique. J’ai regardé la boîte. Fabricant : Inde. J’ai pensé : ‘Ah ouais, c’est bon pour moi, mais ça va pas faire de bien à personne d’autre.’ 😅
Helder Lopes
En Suisse, on a un système hybride : on suit l’EMA mais on adapte les délais de prix. Résultat ? Les génériques arrivent 30 % plus vite qu’en France. Peut-être qu’on a juste appris à simplifier. Pas besoin de 27 systèmes. Un seul bon, c’est suffisant.
Dani Schwander
Les Indiens ? Ils sont pas plus malins. Ils sont juste moins gavés de paperasse. Chez nous, on passe 6 mois à remplir des formulaires pour un médicament qui coûte 2€. Chez eux, ils le font en 2 semaines. C’est pas de la magie, c’est de la logistique. Et on les déteste parce qu’ils nous font honte.
Milad Jawabra
Je viens du Canada, et je peux dire que notre système est plus simple. On a une seule autorisation fédérale, puis on négocie les prix au niveau provincial. Pas de 4 voies. Pas de contradictions entre pays. L’Europe a une chance de faire pareil. Mais elle préfère compliquer. Pourquoi ? Parce que c’est plus rentable pour les bureaucrates.
Julien Doiron
Je me demande si cette réforme n’est pas une manœuvre pour transférer la responsabilité des pénuries vers les pays les plus faibles… Les exigences d’approvisionnement sont floues, et les pays de l’Est sont déjà débordés. Qui va payer pour les stocks de 6 mois ? Les hôpitaux ? Les patients ? C’est un piège. Et je le sens.
Floriane Jacqueneau
En réponse à Julien Doiron : tu as raison, la flou sur les stocks est un piège. Mais ce n’est pas une manœuvre secrète. C’est une conséquence logique d’un système qui n’a jamais voulu harmoniser. Les pays qui ont peur de perdre leur autonomie réglementaire créent des barrières. Ce n’est pas du mal, c’est de la peur.